山西非酒肝原代細胞分離培養
并進質粒抽提。GAG質粒和VSV質粒同樣可以轉化至感受態細菌DH5α中,并進行無內質粒抽提。質粒抽提后冷凍于-20℃保存。2、慢病毒包裝1)使用DMEM完全培養基培養6cm皿HEK293T至匯合度為70~80%。采用opti-MEM和Lipo3000分別轉染含有目的基因的pMSCV-eGFP、VSV、GAG質粒及對照載體,每皿加入脂質體-質粒轉染混懸液按購買脂質體相關說明書操作定量。繼續培養24h。2)24小時后,將培養基更換為新鮮的DMEM完全培養基,放進細胞培養箱繼續培養48~72h。3)48~72h后收集上層培養液,并過μm濾膜,采用ELISA法對所獲得的慢病毒載體進行病毒滴度測定。如不及時使用可以凍存于-80℃。3、慢病毒轉染1)轉染前1天將細胞接種6孔培養板,時細胞的融合率約為50%,前需換液,加入1mLDMEM完全培養基。2)病毒冰浴融化后加入相應體積的病毒液及聚凝胺(Polybrene),混勻后放入37℃孵箱中繼續培養3)4h后補充1mL培養基,14h后換液(24h內換液即可)。4)病毒72h后用倒置顯微鏡觀察熒光,監測效率,出現較多熒光時將等量的轉染細胞和未轉染細胞分別加入等濃度Puromycin(Puromycin或其他篩選濃度需要事先摸索)。5)待未轉染細胞全部死亡并且可觀察到滿意熒光量時,降低Puromycin濃度培養。枯否細胞被命名為肝巨噬細胞,它是我們人體內**的巨噬細胞。山西非酒肝原代細胞分離培養

上期我們主要講解了細胞凋亡的早期檢測方法,這期主要講解一下細胞凋亡晚期中,核酸內切酶(某些Caspase的底物)在核小體之間剪切核DNA,產生大量長度在180-200bp的DNA的片段。1、凋亡DNALadder檢測試劑盒細胞經處理后,采用常規方法分離提純DNA,進行瓊脂糖凝膠和溴化乙啶染色,在凋亡細胞群中可觀察到典型的DNAladder。QuickApoptoticDNALadderDetectionKit可以快速(約1-2小時)、靈敏地檢測凋亡細胞中的DNA的片段。2、TUNEL-basedDNA的片段分析試劑盒細胞凋亡中,染色體DNA雙鏈斷裂或單鏈斷裂而產生大量的粘性3-OH末端,可在脫氧核糖核苷酸末端轉移酶(TdT)的作用下,將脫氧核糖核苷酸和熒光素、過氧化物酶、堿性磷酸酶或生物素形成的衍生物標記到DNA的3-末端,從而可進行凋亡細胞的檢測。Apo-BrdUTM分析使用2種顏色的TUNEL方法用流式細胞儀檢測凋亡細胞中DNA條帶。采用的BrdU比生物素標記的或地高辛(digoxigenylated)標記的方法靈敏度更高。3、Caspase分析試劑和試劑盒Caspase在細胞凋亡中發揮著重要的作用,屬于天冬氨酸蛋白酶。其中caspase-3為關鍵的執行分子,它在凋亡信號傳導的許多途徑中發揮功能。在正常狀態下,caspase家族都以無活性的酶原。青海纖維化原代細胞分離培養評價專業做原代細胞分離的公司。

防止有Matrigel流經上孔而留下殘留膠;3、需要按照細胞的生長速度定時采集圖像,并且對其成管長度、覆蓋面積、成環數和結點進行測量和記錄,并且對其進行統計分析;4、分上下孔的ibidi血管生成載玻片能去除凹液面,使成像效果更好。(Matrigel鋪在下孔,細胞鋪在Matrigel上,上孔充滿培養基)2、數據分析(在特定的時間點采集圖片,并且進行圖像分析(Wimasis全自動分析)測量小管長度,成環數,細胞覆蓋面積和結點。之后在對測量結果進行統計分析以說明實驗結果。)三、實驗具體步驟1、準備基質膠1)實驗前一天將Matrigel置于冰盒中,放入4度冰箱,使膠能過夜緩慢融化。(注意:同樣要準備一些4攝氏度預冷的頭用于吸取Matrigel)。2)開始實驗前,將Matrigel始終保持放在冰盒中。3)打開滅菌包裝,取出ibidi血管生成載玻片。4)每孔中加入10μlMatrigel。注意頭要垂直于內孔的正上方加入Matrigel,防止有Matrigel流經上孔而留下殘留膠。由于Matrigel流動性不強,并且有可能移液不準確,有可能打入10μl的膠,卻不能填滿血管生成載玻片的下孔——這樣,必然會影響到實驗的成像結果。
建議按照2×106每孔的數量將293T細胞均勻鋪入。(2)第二天:在24小時之內,觀察293T細胞的匯合度在90%~95%之間時,向其中加入DNA-脂質體復合體,DNA-脂質體復合體制備方法如下:a)輕輕混勻LipoMax,根據說明書加入相應量于500μlOpti-MEM無血清培養基中,混合均勻并置于室溫5分鐘。b)在500μlOpti-MEM無血清培養基中稀釋DNA,總質量為15μg按照載體質粒:psPAX2:=4:3:1的比例加入DNA。c)將稀釋后的LipoMax和稀釋后的DNA輕輕混勻,常溫靜置20分鐘,形成DNA-LipoMax復合體。(3)將DNA-LipoMax復合體輕柔地滴加至細胞培養皿中,輕輕搖晃培養皿混勻,放入細胞培養箱中培養。(4)病毒收集濃縮病毒:加入DNA-LipoMax復合體48小時后,收集病毒上清,同時加入10ml預溫的293T培養基到細胞培養皿中。將收集到的病毒上清存在4℃冰箱中;收集72小時病毒上清,與48小時病毒上清混在一起。將離心機溫度降溫到4℃,600g,離心5分鐘,去除其中的細胞碎片,上清液經μm濾頭過濾,加入病毒濃縮液,配制濃縮病毒液。將濃縮后的病毒放于4℃冰箱搖床上,旋轉過夜。第二天,4度離心機,3000~4000g離心15分鐘。棄掉上清液,加入1Xpbs或培養基重懸。SD大鼠腎足細胞分離細胞鑒定。
使其形成利于核酸進入的微孔。C.逆轉錄病毒(RNA):通過病毒中膜糖蛋白和宿主細胞表面的受體相互作用進入宿主細胞,之后反轉錄酶啟動合成DNA并隨機整合到宿主基因組中。特點:穩定轉染,可用于難轉染的細胞、原代細胞,體內細胞等,但攜帶基因不能太大。D.腺病毒(雙鏈DNA):先和細胞表面的受體結合,繼而在αV整合素介導下被細胞內吞。特點:可用于難轉染的細胞。2、影響轉染的因素血清:血清影響復合物的形成,降低轉染效率。陽離子脂質體和DNA的量在使用血清時會有所不同,因此想在轉染培養基中加入血清時要進行條件優化。大部分細胞可以在無血清培養基中幾個小時內保持健康。:比如青霉素和鏈霉素,是影響轉染的培養基添加物。這些一般對于真核細胞無毒,但陽離子脂質體試劑增加了細胞的通透性,使可以進入細胞。這降低了細胞的活性,導致轉染效率低。細胞代數:轉染前細胞經過1-2次傳代保證細胞生長旺盛容易轉染,注意貼壁細胞一旦長滿就不好轉染。細胞鋪板密度:一般轉染時,貼壁細胞密度為70%-90%,懸浮細胞密度為2*10^6--4*10^6細胞/ml時效果較好。確保轉染時細胞沒有長滿或處于靜止期。鋪板細胞數目的增加可以增加轉染活性和細胞產量。SD大鼠腎足細胞哪里有賣。山西非酒肝原代細胞分離培養
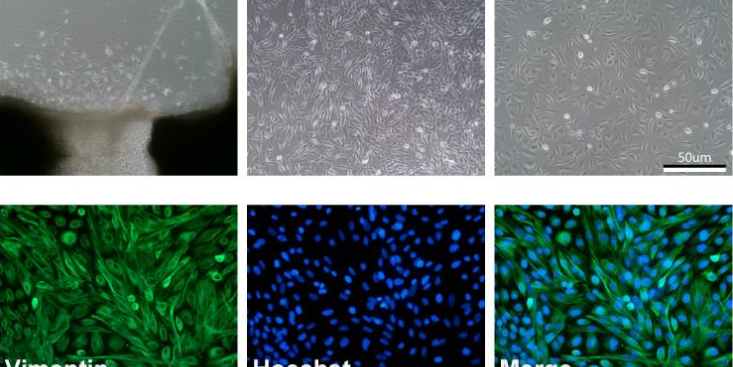
細胞呈長梭形,無橫紋,受自主神經支配,為不隨意肌。山西非酒肝原代細胞分離培養
病毒包裝技術服務病毒包裝技術服務(DH0010)一、服務介紹重組病毒以其高效和多樣性,是非常有效的將外源基因導入細胞的方法。慢病毒腺病毒腺病毒相關表達特點穩定表達瞬時表達瞬時表達是否整合到基因組是否否免疫原性弱強弱病毒**力高很高高注:高濃度時可能有極少量整合發生。二、服務內容及價格服務編號服務內容服務價格服務周期(工作日)DH0010-1慢病毒包裝咨詢咨詢DH0010-2腺病毒包裝咨詢咨詢DH0010-3腺相關病毒包裝咨詢咨詢注:根據客戶實驗需要靈活定制病毒載體包裝服務,滿足客戶研究的多種需要。三、客戶提供1.客戶需提供生長狀態良好的細胞株及其他**(處理細胞**)實驗材料(默認由我方提供)2.靶基因信息。3.實驗前,客戶需告知具體的細胞處理方式及詳細要求。注:若有特殊處理的樣品,請盡可能出示相關材料(具有保密性的材料除外)。四、服務流程1)根據目的基因相關信息(序列,序列號等),對每條目的設計3條mRNA序列,其中一條序列為陰性對照組;2)根據設計好的mRNA序列,設計,合成兩條互補的短片段的DNA;3)對合成好的DNA進行片段稀釋,退火,連接到線形化處理過的載體,轉化,涂平板,過夜培養;4)通過PCR的方法篩選陽性菌落,進行測序。山西非酒肝原代細胞分離培養
- 貴州Balbc裸鼠原代細胞分離培養原理 2025-11-06
- 湖北生殖原代細胞分離培養評價 2025-11-06
- 寧夏皮膚原代細胞分離培養說明書 2025-11-06
- 遼寧C57小鼠原代細胞分離培養供應商 2025-11-06
- 海南如何原代細胞分離培養評價 2025-11-06
- 山西非酒肝原代細胞分離培養 2025-11-06
- 為什么原代細胞分離培養購買 2025-11-06
- 上海纖維化原代細胞分離培養優勢 2025-11-06
- 云南呼吸原代細胞分離培養公司 2025-11-06
- 浙江正規原代細胞分離培養廠家 2025-11-06
- 甘肅數字孿生智慧園區 2025-11-06
- 云南共享洗烘企業合作 2025-11-06
- 美國功能性食品展主辦方 2025-11-06
- 湘西詐騙案是什么意思 2025-11-06
- 六合區運營懸掛式七氟丙烷滅火消防裝置 2025-11-06
- 甘肅市場營銷策劃 2025-11-06
- 崇明區一站式房屋檢測鑒定概況 2025-11-06
- 小鎮規劃建設 2025-11-06
- 深圳交付策劃房屋查驗咨詢師 2025-11-06
- 蘇州智能化高新企業認證費用是多少 2025-11-06